A 2019-es új koronavírus vagy koronavírus-betegség (COVID-19) kitöréséről azt állítják, hogy 2020 eleje óta fenyegetést jelent az egész világra. A tudósok éjjel-nappal azon dolgoznak, hogy megértsék a COVID-19 eredetét. Talán már hallottuk a hírt, hogy közzétették a SARS-CoV-2 feltételezett genomját[1]. Hogyan is történik tehát pontosan a SARS-CoV-2 teljes genomjának azonosítása?
Ez a cikk tisztán technikai módon elmagyarázza, hogyan történik a folyamat. Ennek eredményeképp tisztában leszünk vele, hogy ez a módszer nem alkalmas sem a mintában lévő nukleinsav eredetének meghatározására, sem a genom pontos meghatározására. Különféle algoritmusokat használnak, amelyek emberi utasítások alapján próbálnak egy fiktív konstrukciót javasolni, amely aztán a minden további folyamathoz sablonként szolgál. Azonban minden egyes szekvenálás olyan eredményeket ad, amelyek eltérnek az előző szekvenálástól, és ezeket az eltéréseket gyakran mutációkként adják el[2], amelyek egyszerűen a genomanalízis eszközök által végzett számítások pontatlanságai.
Tekintettel arra, hogy tiszta tenyészetben soha nem izoláltak vírusosnak vélt szerkezetet és így abból nem is tudtak közvetlenül biokémiai jellemzést végezni, hanem minden szekvenálás olyan genetikai anyag keverékén alapul, amelynek eredete nem állapítható meg, minden számítógéppel előállított konstrukciót puszta spekulációnak kell tekinteni.
Előzetesen egy apró, de nem kevésbé fontos információként, amely tökéletesen egyértelművé teszi számunkra, hogy a sorbarendezés/összeállítás (alignment/assembly) technikai folyamata (ahogyan azt a cikkben rögtön megmagyarázzuk) tisztán fiktív és manipulatív:
A virológusok 70%-a nem közli, hogy melyik összeállítási technikát alkalmazza!
És a kimondatlan tény az, hogy a virológusok 100%-a nem számol be arról, hogy a kiszámított (gépen előállított) “vírus” genom hány százaléka változott meg, amíg azt kész genomként nem publikálták.
A kínai Fan Wu et. al [1] által közétett és letölthető genom adatok semmiképp nem nyers, hanem manipulált adatok! Az emberi szekvenciák 80%-a eltűnt, és mindenekelőtt: mindent kiszűrtek, amit máshol „vírusként” publikáltak! Ez még nehezebbé teszi egy új, betegséget okozó vírus állítólagos felfedezésének reprodukálhatóságát, hiszen hiába keresünk kontrollkísérleteket a publikációkban.
Lássunk most hozzá a technikai genomelemzéshez.
Genom
A genom a genetikai anyag teljessége, beleértve a szervezet összes génjét. A genom minden olyan információt tartalmaz az élőlényről, amely szükséges a felépítéséhez és fenntartásához.
- A betegséget okozó vírusokat az definiálja, hogy szekvenciájuk (genomjuk) egyedi, egészséges szervezetekben nem fordul elő.
- Ahhoz, hogy egy vírus genetikai anyagának jelenlétét igazolni és meghatározni lehessen, ezt a vírust izolálni kell és tiszta formájában elérhetővé kell tenni a gondolkodás törvényszerűségei és a logika alapvető szabályai szerint, melyek minden tudománynak alapvető fetlételei. Így lesz csak elkerülhető, hogy a sejt saját génszekvenciáit ne értelmezzük félre egy vírus összetevőjeként .
- Egy genetikai anyag szekvenciájának meghatározása csak akkor lehetséges, ha az DNS formában van.
Szekvenálás
Hogyan olvashatók ki a genomban jelenlévő információk? Itt jön képbe a szekvenálás.
A szekvenálást egy szervezet egyes génjeinek, egész kromoszómáinak vagy egész genomjai szekvenciájának meghatározására használják.

1. ábra: Egy PacBio szekvenálógép. A PacBio egy harmadik generációs szekvenálási technológia, amely hosszú leolvasásokat eredményez. KENNETH RODRIGUES képe a Pixabayről (CC0)
A szekvenálógépeknek nevezett speciális gépeket arra használják, hogy a genomból rövid, véletlenszerű szekvenciákat nyerjenek ki, amelyek minket érdekelnek. A jelenlegi szekvenálási technológiák nem képesek a teljes genomot egyszerre kiolvasni. A kis darabokat átlagosan 50-300 bázis (újgenerációs szekvenálás/rövid olvasás) vagy 10 000-20 000 bázis (harmadik generációs szekvenálás/hosszú olvasás) közötti hosszúsággal olvassák le, az alkalmazott technológiától függően.
Ezeket a rövid darabokat olvasásnak (read) nevezzük.
- Az örökítőanyag jelenlétét és hosszát úgy határozzuk meg, hogy elektromos térben hosszirányban szétválasztjuk. A rövid darabok gyorsabban, a hosszabbak lassabban vándorolnak. Ugyanakkor a vizsgálandó genetikai anyag hosszának meghatározásához különböző hosszúságú, ismert hosszúságú genetikai anyagdarabokat adnak hozzá. A genetikai anyag hosszának kimutatására és meghatározására szolgáló megbízható szabványos technikát “gélelektroforézisnek” nevezik.
- Ha egy bizonyos genetikai anyag koncentrációja túl alacsony ahhoz, hogy a “gélelektroforézis” technikájával kimutatható legyen, akkor a DNS korlátlan szaporításának technikájával, az úgynevezett polimeráz láncreakcióval (PCR) tetszés szerint megsokszorozható. Ily módon a gélelektroforézis során a nem kimutatható DNS láthatóvá tehető. Ez az előfeltétele annak, hogy a genetikai anyag hozzáférhetővé váljon a további vizsgálatok számára, különösen a hosszának és szekvenciájának későbbi, döntő meghatározásához. Ezt a módszert PCR-ként is rövidítik.
Ha részletesebb információt keresünk arról, hogy pontosan hogyan működik a vírusgenomok szekvenálása klinikai mintákból, a következő cikkekben találja meg.
2. Klinikai mintákból származó vírusok specifikus befogása és teljes genom szekvenálása
Genom-összerakás (Genom-assembly)
Amint a genom kis részei rendelkezésre állnak, az átfedési információik alapján össze kell őket kombinálnunk, és össze kell állítanunk a teljes genomot. Ezt a folyamatot nevezik összeállításnak, összerakásnak (assembly), és egy kirakós játék megoldásához hasonlítható. A leolvasások átfedésük alapján történő összerakására speciális, asszemblereknek nevezett szoftvereszközöket használnak. Ez a kontigoknak nevezett folyamatos karakterláncok létrehozására szolgál. Ezek a kontigok lehetnek a teljes genom vagy annak csak töredékei (ahogy a 2. ábrán látható). Ami itt fontos, hogy az anyag forrása nem számít.
A kínai virológusok nem végeztek ellenőrző kísérleteket, hogy kizárják annak lehetőségét, hogy lehetséges pontosan ugyanannak a vírusgenomnak a kreálása rövid RNS-töredékekből
- egészséges személy tüdőváladékából származó emberi/mikrobiológiai RNS,
- egy másfajta tüdőbetegségben szenvedő személy,
- negatív SARS-CoV-2 tesztet felmutató. személy
- vagy a SARS-CoV-2 vírus felfedezése előtt eltett mintákból származó RNS esetében.
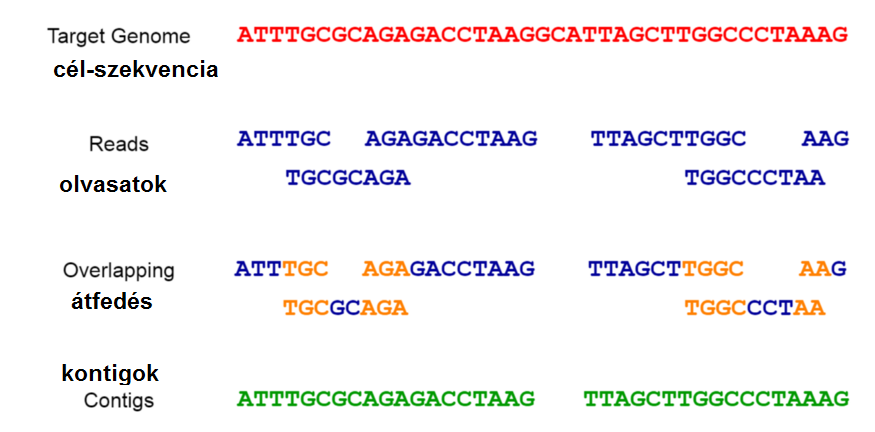
2. ábra Szekvenálás és összerakás
Az asszemblálók két kategóriába sorolhatók,
1, de novo asszemblerek: referencia genomok használata nélkül asszemblereznek (pl.: SPAdes, SGA, MEGAHIT, Velvet, Canu és Flye).
2, Referencia-vezérelt asszemblerek: szekvenciák referencia genomokhoz való hozzárendelésével állítják össze.
Az összeszerelők két fő típusa
A bioinformatikai szakirodalomban két fő típusát találjuk az asszemblereknek. Az első típus az átfedéses elrendezési konszenzus (Overlap-Layout-Consenses; OLC) módszer. Az OLC-módszerben először meghatározzuk a leolvasások közötti átfedéseket. Ezután az összes olvasás és átfedés egy gráf formájában kerül elrendezésre. Végül azonosítjuk a “konszenzus szekvenciát”. Az SGA egy népszerű eszköz, amely az OLC-módszeren alapul.
A második típusú összeszerelő a de-Bruijn-gráf (DBG) módszer[3]. Ahelyett, hogy a teljes leolvasott adatot használná, a DBG-módszer a leolvasott adatot még rövidebb fragmentumokra, úgynevezett k-merekre bontja (k hosszúságúak), majd az összes k-mer felhasználásával létrehoz egy de-Bruijn-gráfot. Végül a genomszekvenciákat a de-Bruijn-gráf alapján következtetjük le. Az SPAdes egy népszerű, DBG-módszeren alapuló asszemblerező.
Mi romolhat el a genom összerakása során?
A genomok olyan nukleinsavmintákat tartalmaznak, amelyek a genomban többször is előfordulnak. Ezeket a struktúrákat ismétlődéseknek nevezzük. Ezek az ismétlődések megnehezíthetik az összeállítási folyamatot, és kétértelműségekhez vezethetnek.
Nem tudjuk garantálni, hogy a szekvenáló a teljes genomot lefedő leolvasásokat (read-okat) tud előállítani. Előfordulhat, hogy a szekvenáló kihagyja a genom bizonyos részeit, és nincsenek olyan read-ok, amelyek az adott régiót lefednék. Ez hatással lesz az összeállítási folyamatra, és ezek a kihagyott régiók nem lesznek jelen a végső összeállításban.
A genom-összeszerelőknek foglalkozniuk kell ezekkel a kihívásokkal, és meg kell próbálniuk minimalizálni az összeállítás során okozott hibákat.
Hogyan értékelik ki a kapott részeredményeket?
A nukleinsavcsoportok kiértékelése nagyon fontos, mivel el kell döntenünk, hogy a kapott csoport megfelel-e a szabályoknak. Az egyik legismertebb és leggyakrabban használt összeállítási értékelő eszköz a QUAST. Az alábbiakban bemutatunk néhány kritériumot az összeállítások értékeléséhez.
- N50: az összeállítás (nukleinsavcsoport) teljes hosszának 50%-ához szükséges minimális kontighossz.
- L50: az N50-nél hosszabb kontigok száma
- NG50: a referencia genom hosszának 50 %-ának lefedéséhez szükséges minimális kontighossz.
- LG50: az NG50-nél hosszabb kontigok száma
- NA50: az összehangolt blokkok minimális hossza, amely az összeállítás teljes hosszának 50 %-ához szükséges.
- LA50: az NA50-nél hosszabb kontigok száma
- Genom százalékos aránya (%): a referencia genommal megegyező bázisok százalékos aránya.
Piszkoljuk be kezünket
Kezdjünk hozzá a kísérletezéshez. Az SPAdes asszemblert fogom használni a szekvenált páciensmintákból nyert olvasatok összerakásához. Az SPAdes újgenerációs szekvenálási leolvasásokat használ. A QUAST-ot ingyenesen is letölthetjük. A kódot és a bináris állományokat a megfelelő honlapokról szerezheti be, és futtathatja ezeket az eszközöket (tool-okat).
Írjuk be a következő parancsokat, és ellenőrizzük, hogy az eszközök megfelelően működnek-e.
 <your_path_to>/SPAdes-3.13.1/bin/spades.py -h
<your_path_to>/SPAdes-3.13.1/bin/spades.py -h
<your_path_to>/quast-5.0.2/quast.py -h
Az adatok letöltése
Feltételezem, Ön tudja, hogyan tölthet le adatokat a Nemzeti Biotechnológiai Információs Központból (NBCI). Ha nem, akkor erre a linkre hivatkozhat.
A kísérleteinkhez szükséges leolvasások letölthetők az NCBI-ból az SRX7636886-os NCBI-belépési számmal. Letölthetjük az SRR10971381 futtatást, amely egy Illumina MiniSeq futtatásból származó leolvasásokat tartalmaz. Győződjünk meg róla, hogy az adatok FASTQ formátumban kerülnek letöltiésre. A letöltött fájl “sra_data.fastq.gz” néven található. A FASTQ-fájlt a gunzip programmal lehet kbontani.
A kibontás után futtassuk a következő bash parancsot, hogy a program megszámolja az olvasatok számát az adathalmazunkban. Látni fogja, hogy 56.565.928 olvasás van.

grep ‘^@’ sra_data.fastq | wc -l
A nyilvánosan elérhető SARS Cov-2 teljes genom[1] letölthető az NCBI-ről a GenBank hozzáférési számmal MN908947 (Kérjük, ne feledje, hogy ez nem a valódi nyers adat). Egy FASTA formátumú fájlt fog látni. Ez lesz a referencia genomunk. Megjegyzendő, hogy az át lett nevezve “MN908947.fasta”-ra.
Összeállítás
Állítsuk össze a COVID-19 olvasatait. Futtassa a következő parancsot a leolvasott adatok SPAdes segítségével történő összeállításához. A tömörített .gz fájlt közvetlenül átadhatjuk az SPAdes-nek.
<your_path_to>/SPAdes-3.13.1/bin/spades.py –12 sra_data.fastq.gz -o Output -t 8
A cikkhez az általános SPAdes asszemblert használtuk demonstrációként. Mivel azonban a reads adathalmaz RNS-Seq adatokból áll, jobb, ha a SPAdes-ben a “-rna” opciót használjuk.
A kimeneti mappában látni fogunk egy “contigs.fasta” nevű fájlt, amely a véglegesen összeállított kontigokat tartalmazza.
Az összeállítási eredmények értékelése
Futtassuk a QUAST-ot a nunkleinsavcsoportokra a következő paranccsal.
![]()
<your_path_to>/quast-5.0.2/quast.py Output/contigs.fasta-l SPAdes_assembly -r MN908947.fasta -o quastResult
Az értékelési eredmény megjelenítése
Amikor a QUAST befejeződött, beléphetünk a “quastResult” mappába, és megtekinthetjük az értékelési eredményeket. A QUAST-jelentést a report.html fájl webböngészőben történő megnyitásával tekinthető meg. Egy, a 3. ábrán láthatóhoz hasonló jelentés jelenik meg. A “részletes jelentés” gombra kattintva további információkat kaphatunk, például NG50 és LG50.
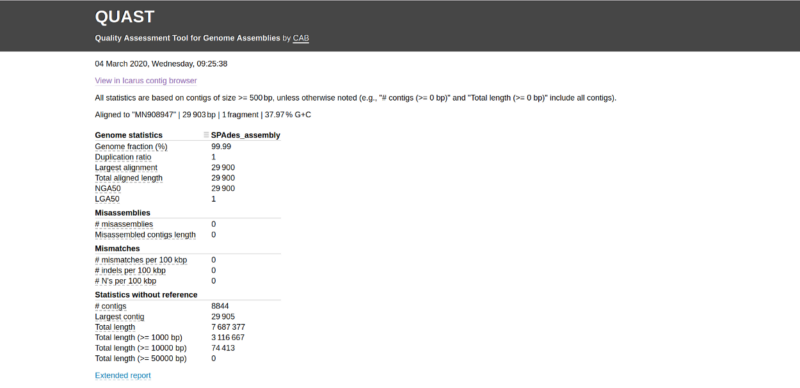
3. ábra: QUAST-jelentés
Megvizsgálhatjuk a különböző értékelési kritériumok értékeit, például az NG50, NA50 genom arányok, a félresikerült összeállításokat és a kontigok számát. A referencia genomhoz történő “Contig sorrendbe állítást (Contig-Alignment)” az Icarus contig böngésző segítségével is megtekinthetjük [kattintsunk a “View in Icarus contig browser” (Nézet az Icarus contig böngészőben) gombra], amint az a 4. ábrán látható.
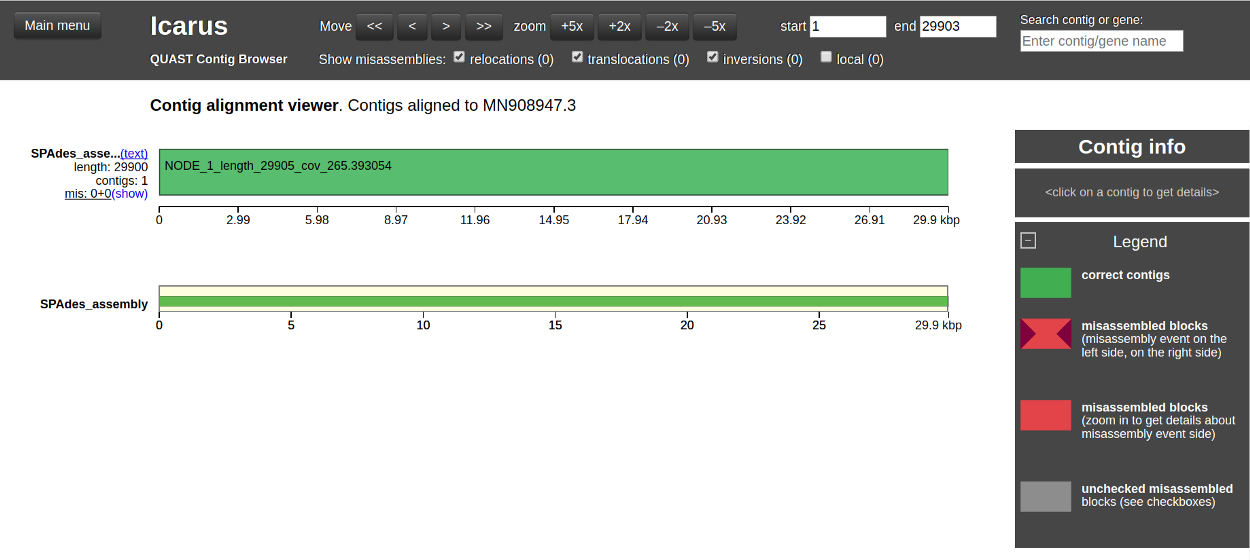
4. ábra: QUAST jelentés
Az Icarus contig böngészőben láthatjuk, hogy a “NODE_1” nevű kontig nagyon jól illeszkedik a COVID-19 referencia genomjához. Genomtartalma 99,99% (amint az a 3. ábrán látható). Ezen túlmenően a 29 900 bázispár teljes igazított hossza nagyon közel van a referencia genom hosszához, amely 29 903 bázispár.
Az összerakási diagram vizualizálása
Van egy Bandage nevű eszköz, amely lehetővé teszi az összeállítási gráf megjelenítését. Letöltheti az előre lefordított bináris fájlokat a honlapjukról, és futtathatja a segédprogramot.
A SPAdes kimeneti mappájában található gráf-fájlt betölthetjük a Bandage-ba “assembly_graph_with_scaffolds.gfa”
válasszuk ki a gfa-fájlt, nyissuk meg,
és a “Draw graph” gombra kattintva az 5. ábrán látható módon megjeleníthetjük.
Vegyük észre, hogy az 5. ábrán az első szegmenssor közepén lévő hosszú, zöld színű, ívelt szegmens megfelel az SPAdes összeállításunk NODE_1-jének.

5. ábra: A COVID-19 read adathalmaz SPAdes összeállítási grafikonjának egy része Bandage segítségével megjelenítve.
Gyakorlati példa a towardsdatascience oldalról
Hogyan találták meg először a SARS-CoV-2 genomját?
Most, hogy a SARS-CoV-2 referencia genomja rendelkezésre áll, ki tudjuk értékelni az összeállításunkat. Kezdetben azonban nem létezett pontos SARS-CoV-2 referencia genom. Mit tettek a tudósok tehát, hogy megállapítsák? Amint az ebben a cikkben kifejtésre kerül, a vírusgenomok elemzése a metagenomikához tartozik, és erre számos technika létezik. Elemezték a kontigok lefedettségét (a kontigban minden egyes bázispozíciót lefedő olvasatok átlagos száma), és összehasonlították a denevér SARS-szerű coronavírus (CoV) izolátummal – denevér SL-CoVZC45 (GenBank hozzáférési szám. MG772933) [4] Az eredmények azt mutatták, hogy a leghosszabb összeállított kontigjuk “magas” lefedettséggel rendelkezett (a mi összeállításunkból látható, hogy a NODE_1 szintén magas lefedettséggel rendelkezik), és nagyon szoros rokonságban állt a denevér SL-CoVZC45-vel.
A kínai virológusok még arra is kifejezetten rámutatnak, hogy a konstruált genomszál akár 90%-os hasonlóságot mutat a denevérek ártalmatlan és állítólagos koronavírusainak évtizedek óta ismert genomszálaival.

Ez a 90%-os hasonlóság abból adódik, hogy a számos nagyon rövid génszekvencia összehangolásához pontosan egy ártalmatlan denevér “koronavírus” “genomját” használták sablonként. Meg kell jegyezni, hogy ez a denevérgenom, mint a patogén “vírusok” összes “genomja”, csak számítógépes program eredménye, azaz egy gondolati konstrukció, nagyon rövid endogén (az emberi sejt részeként meglévő) génszekvenciákból és/vagy számos mikroba géntöredékeiből, amelyek a valóságban soha nem kerültek elő teljes örökítőszálként, és sehol sem jelennek meg teljes “vírusgenomként” a tudományos irodalomban.
Így a 2020.01.24-i tanulmány a Discussion – Megvitatás alatt megállapítja:
„Our study does not fulfill Koch’s postulates“
“Tanulmányunk nem teljesíti Koch posztulátumait”.
Itt fontos tudni, hogy nem ismert, hogy a kiszámított “vírus” genom hány százalékát változtatták meg, mielőtt kész genomként publikálták volna.
A Fan Wu és munkatársai által a Nature, Vol 579, 2020.2.3. számában megjelentetett publikáció, amelyben először mutatták be a SARS-CoV-2 genomját (teljes genomszálát), és amely minden további sorrendbe állítás (alignment) mintájává vált, azt mutatta, hogy egyértelműen egy beteg személy hörgőváladékából (BALF) nyert összes RNS-t felhasználták, anélkül, hogy előtte izolálták vagy dúsították volna a virális struktúrákat vagy nukleinsavakat.
Ebben a publikációban Zhang professzor leírja, hogy egy adott genomszekvencia (ártalmatlan denevérkoronavírus) alapján hét különböző, igen összetett módszerrel, köztük statisztikai módszerekkel, hogyan számolt ki egy 29 903 nukleotidból álló genomot rövid, mindössze 21 és 25 nukleotid hosszúságú génszakaszok alapján (ezek az alkalmazott alignment programok – Megahit és a Trinity – alapértelmezett paraméterei).
Ezt az RNS-t ezután cDNS-sé alakították át (PCR), és a mindössze 150 nukleotid hosszúságú molekulákat szekvenálták, hogy pusztán számítással megépítsék a kb. 30 000 nukleotid hosszúságú teljes genomot.
Feltételezi – anélkül, hogy ezt kifejezetten kimondaná -, hogy azok a rövid szekvenciák, amelyekből javasolja a SARS-CoV-2 vírus genomjának szekvenciáját, vírusos jellegűek, mert kizárja azokat a hosszabb szekvenciákat, amelyek a rövid 21 és 25 bázispárból álló szakaszok átfedéséből (= contigs) származnak, és amelyek az emberi szekvenciákkal mutatnak hasonlóságot.
Egyszerűen fogalmazva ez azt jelenti:
Mivel a keverékből az általunk “ismert” emberi szekvenciákat szoftveres úton eltávolították a genetikai anyag keverékéből, a megmaradt szekvenciákat – amelyek a zavaros virológiai kényszeres gondolkodás produktumai (amelyért 1954-ben John Franklin Enders Nobel-díjat kapott, aki elméletét csupáncsak hipotézisként állította fel,)[5], – vírusos természetűnek feltételezik.
Az, hogy a teljes genom hány százaléka tartalmaz hiányzó helyeket (gap) (1% vagy majdnem az összes??), nem közlik.
Logikus következmény:
Amit itt mindenféle lépésekben mesterségesen létrehoztak, mindezt pusztán elfogadott (elhitt), de soha nem igazolt “feltételezések” alapján, annak abszolút NINCS köze a valósághoz! A szekvenálási módszer nem tudja megmondani, hogy a sok számítási lépéssel létrehozott genom milyen feltételezett forrásból származik, és azt sem, hogy egyáltalán tisztán vírusos jellegű-e. Ez legfeljebb egy olyan eszköz, amely különböző algoritmusok és úgynevezett hiánypótló programok segítségével (a genomkonstrukcióban lévő lyukakat kitölti) sok nagyon rövid génszekvenciából egy új, fiktív, konstruált genomszálat hoz létre. Több mint arcátlanság azt állítani, hogy pontosan ez a számítógépes konstrukció (a genom) – amelyet egy beteg BALF (hörgőváladék) mintájából vettek – vírusos jellegű, anélkül, hogy izoláltak volna egy bizonyos struktúrát, csak azért, mert a beteg tüneteket mutatott.
A bioinformatikusokat nem érdekli a génszekvencia forrása.
Amikor az influenzavírusok genetikai szálainak ötlete először született meg, csirkeembriókat mechanikusan megsebeztek és megmérgeztek, majd az elhaló szövetek rövid nukleinsav-szekvenciáiból aprólékos kézi munkával modellt készítettek. Ezt ma már számítógépes programok végzik, amelyeket olyan szekvenciákkal táplálnak, amelyeket a virológusok “vírusosnak” tartanak. Az, hogy ezek a szekvenciák valójában honnan származnak, lényegtelen a bioinformatikusok számára, akik a fiktív vírusok genomjait “sorba rendezéssel, összehangolással (alignment)” hozzák létre. Erwin Chargaff egyébként már 1976-ban figyelmeztetett erre a fejleményre “Hérakleitosz tüze” című könyvében.
Források
[1] F. Wu, S. Zhao, B. Yu et al. A humán légúti megbetegedésekkel összefüggésbe hozott új koronavírus Kínában. Nature (2020). https://doi.org/10.1038/s41586-020-2008-3
[2] Az Angliából származó állítólagos SARS-CoV-2 mutáció csak egy becsomagolt svindli.
[3] Zhenyu Li et al. Comparison of the two major classes of assembly algorithms: overlap-layout-consensus and de-bruijn-graph, Briefings in Functional Genomics, Volume 11, Issue 1, January 2012, Pages 25-37. https://doi.org/10.1093/bfgp/elr035. https://doi.org/10.1093/bfgp/elr035
[4] Fan Wu: Egy új koronavírus, amely emberi légúti megbetegedésekkel áll kapcsolatban Kínában.
 A WHCV vírusgenom-szerveződését a Betacoronavirus nemzetség két reprezentatív tagjával való szekvencia-illesztéssel határozták meg: egy emberekkel kapcsolatos koronavírussal (SARS-CoV Tor2, GenBank hozzáférési szám: AY274119) és egy denevérekkel kapcsolatos koronavírussal (denevér SL-CoVZC45, GenBank hozzáférési szám: MG772933).
A WHCV vírusgenom-szerveződését a Betacoronavirus nemzetség két reprezentatív tagjával való szekvencia-illesztéssel határozták meg: egy emberekkel kapcsolatos koronavírussal (SARS-CoV Tor2, GenBank hozzáférési szám: AY274119) és egy denevérekkel kapcsolatos koronavírussal (denevér SL-CoVZC45, GenBank hozzáférési szám: MG772933).
[5] Power work – a vírus cáfolatának alapjai
[6] Jang-il Sohn és Jin-Wu Nam. A de novo teljes génállomány-összeállítás jelene és jövője. Briefings in Bioinformatics, Volume 19, Issue 1, January 2018, Pages 23-40. https://doi.org/10.1093/bib/bbw096.
[7] S. Heerema és C. Dekker. Grafén nano-eszközök DNS-szekvenáláshoz. Nature Nanotech 11, 127-136 (2016). https://doi.org/10.1038/nnano.2015.307
Az írás először Samuel Eckert német vállalkozó honalapján jelent meg 2021. májusában. Szerzőt nem ad meg, így őt tekintjük szerzőnek.
2022. április
Közzéteszi: Király József

Döbbenet…
És most van fordítás alatt az [5] alatti mű.